Ver categorias
Explorar
Fiverr Pro
Português
$
USD
Apaixonado por aprender
Sobre este serviço
Você está trabalhando em um sistema proteína-ligante e precisa de simulações de dinâmica molecular confiáveis e bem analisadas para sua pesquisa ou publicação? Ofereço serviços completos de simulação MD com AMBER, desde a preparação do sistema até figuras prontas para publicação, usando o mesmo pipeline utilizado em pesquisas revisadas por pares na descoberta de medicamentos computacional.
Sou pesquisador de biologia computacional com experiência prática em AMBER/AmberTools, cpptraj e fluxos de análise baseados em Python. Meu trabalho apoiou diretamente manuscritos enviados a periódicos revisados por pares em farmacologia computacional.
O que entrego
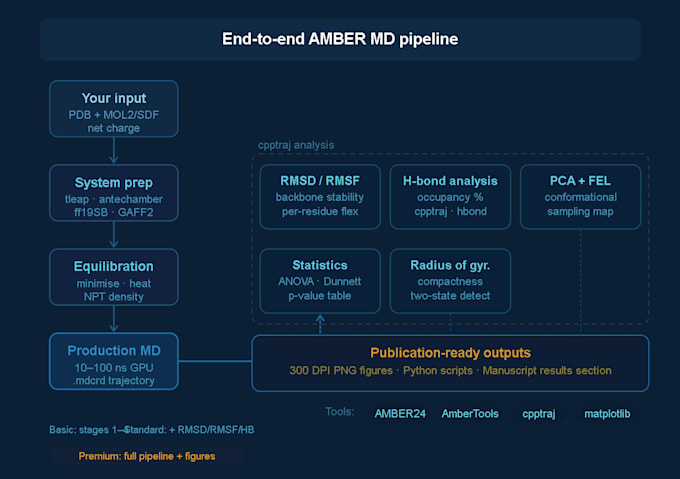
Configuração do sistema
Preparação da estrutura da proteína e atribuição do estado de próton
Parametrização do ligante usando antechamber
Solução (caixa de água TIP3P ou OPC), adição de contraíons
Atribuição do campo de força: ff19SB (proteína), GAFF2 (molécula pequena)
Minimização, aquecimento e equalização NPT/NVT
Execução de MD de produção
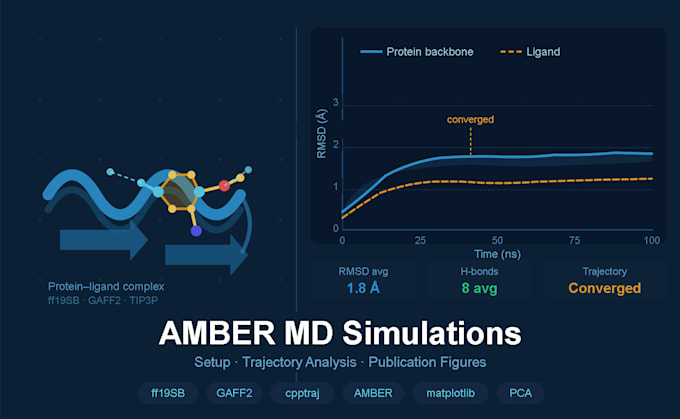
Trajetórias de produção de 10, 50 ou 100 ns (dependente do pacote)
Arquivos completos de trajetória .mdcrd entregues
Análise de trajetória (Padrão & Premium)
Figuras prontas para publicações completas
Por que trabalhar comigo?
Experiência real com amber, não uma ferramenta online ou wrapper na nuvem.